

Towards an Exact Description of Electronic Wavefunctions in Real Solids
Category: Scientific HighlightsPUBLISHED IN "NATURE"
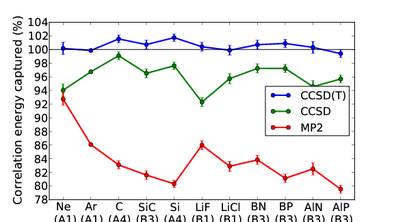
ABSTRACT / ENGLISH - The properties of all materials arise largely from the quantum mechanics of their constituent electrons under the influence of the electric field of the nuclei. The solution of the underlying many-electron Schrödinger equation is a ‘non-polynomial hard’ problem, owing to the complex interplay of kinetic energy, electron–electron repulsion and the Pauli exclusion principle. The dominant computational method for describing such systems has been density functional theory. Quantum-chemical methods—based on an explicit ansatz for the many-electron wavefunctions and, hence, potentially more accurate—have not been fully explored in the solid state owing to their computational complexity, which ranges from strongly exponential to high-order polynomial in system size. Here we report the application of an exact technique, full configuration interaction quantum Monte Carlo, to a variety of real solids, providing reference many-electron energies that are used to benchmark rigorously the standard hierarchy of quantum-chemical techniques, up to the ‘gold standard’ coupled-cluster ansatz, including single, double and perturbative triple particle–hole excitation operators. We show the errors in cohesive energies predicted by this method to be small, indicating the potential of this computationally polynomial scaling technique to tackle current solid-state problems.
Figure 5: Relative errors of the hierarchy of quantumchemical
Published in "Nature": Nature 2012 | ||
Project Part: | P02 Towards Exact Correlations in Extended Systems / Georg Kresse | |
External Link: | ||
External Link: | Computational Materials Physics / Faculty of Physics / University of Vienna | |
External Link: | University of Vienna / Öffentlichkeitsarbeit und Veranstaltungsmanagement | |
Sensengasse 8/12
A-1090 Vienna
AUSTRIA
T: +43-1-4277-51401
F: +43-1-4277-9514